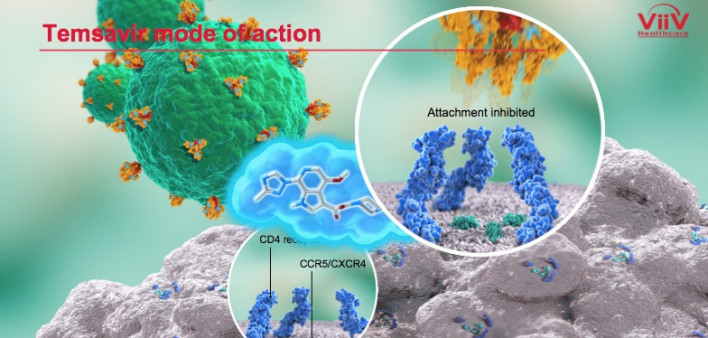
ViiV Healthcare has filed a new drug application with the Food and Drug Administration (FDA) for approval of the first-in-class attachment inhibitor fostemsavir for the treatment of HIV. The new antiretroviral (ARV) is meant for people who have considerable experience with ARVs, have multidrug-resistant virus and cannot assemble a regimen that fully suppresses their virus because of drug resistance, intolerance to certain medications or safety considerations.
The FDA application is backed by 96-week data from the partially randomized, double-blind, placebo-controlled international Phase III BRIGHTE trial. Findings from the trial were presented in July at the 10th International AIDS Society Conference on HIV Science in Mexico City.
The trial included 371 people with HIV who had a high level of experience with ARVs and multidrug-resistant virus. They were required to have a viral load of at least 400 and no more than two remaining classes of ARVs available to them as a result of drug resistance, drug intolerability, contraindication (meaning a drug should not be used for safety concerns, such as a potential interaction with another medication) or other safety concerns.
The participants were placed into either a randomized or a nonrandomized cohort.
The 272 people placed in the randomized cohort had one but no more than two fully active and available ARVs at the study’s outset that could be combined to create an effective background regimen for use along with fostemsavir. The 99 people in the nonrandomized cohort had no fully active approved ARVs.
For the first eight days of the study, 203 individuals in the randomized cohort received twice-daily fostemsavir on a blinded basis (meaning they did not know whether they were receiving the drug or a placebo) while 69 people received a placebo. All of them kept taking their existing failing ARV regimen during this introductory period. Starting on day 9, all of those in the randomized cohort received open-label fostemsavir (meaning they all knew they were taking it) plus a background ARV regimen optimized to work as well as possible.
All the nonrandomized participants received open-label fostemsavir plus an optimized background ARV regimen—one that could include other investigational HIV medications—starting on day 1.
By day 8, those in the randomized cohort who received fostemsavir had experienced an average 0.79 log10 (84%) decline in their viral load, compared with a 0.17 log10 (32%) decline in the placebo group. The difference between these two figures was statistically significant, meaning it is unlikely to have been driven by chance.
At weeks 24, 48 and 96, the proportion of those in the randomized cohort who had a viral load below 40 was 53%, 54% and 60%, respectively. The average change in CD4 cell count from the study’s baseline level was an average increase of 90 at week 24, 139 at week 48 and 205 at week 96. The most common adverse reactions, experienced by at least 5% of the participants, were nausea and diarrhea. At week 96, 5% of the randomized cohort and 12% of the nonrandomized cohort discontinued fostemsavir because of an adverse health event.
Fostemsavir has been granted FDA fast track and breakthrough therapy designations, which means the agency will facilitate and expedite the development and review of the ARV. The fast track designation is granted to therapies that address an unmet medical need in the treatment of a serious or life-threatening condition. To receive a breakthrough therapy designation, a treatment must have demonstrated that it holds the potential for substantial improvement over currently available treatments.
To read a press release about the new drug application, click here.



Comments
Comments